Differences in Indian & Global Pharmacovigilance Requirement | Comparison of USA, India, European Pharmacovigilance System
Pharmacovigilance in India
Indian pharmaceutical industries are estimated to account for about 3.5% in value of the international pharmaceutical industry. By 2020, it is expected to grow to US$55 billion and US$100 billion by 2025, thus evolving as the sixth largest pharmaceutical market globally.
Branded generics constitutes about 80% of the market share, and New Chemical Entities (NCEs) are also being familiarized in the nation, also subsequently emerging as a significant crossroad for clinical
research as well as various outsourcing programs.
PV in the USA
In the USA, the FDA had introduced the FDA Adverse Event Reporting System (FAERS; previously known as Adverse Event Reporting System), to support postmarket surveillance, which allows manufacturers, medical management professionals, and subjects to present adverse event reports. Information about adverse happenings, medication errors, product quality complaints, and patient demographics is available in the database.
MedWatch is a web-based reporting system of FDA that lets patients and health care professionals willingly declare severe detrimental events and other unfavorable problems that they
suspect are linked with the use of an FDA-regulated product.
The adverse events can be reported in detail to the manufacturers or the FDA. MedWatch provides information on voluntary and mandatory reporting. The ADRs are reported online through 2 form 3500A or 3500B, which are reviewed by the Centre for Drugs Evaluation and Research or the Centre for
Biologics Evaluation.
The FDA introduced the Sentinel Initiative under the US FDA Amendments Act (2007) for the submission of adverse events mandatory for the manufacturers.
PV in Europe
EudraVigilance is the framework for assembling, managing, and examining alleged ADRs of drugs approved within the European Economic Area. The European Medical Agency was set up in 1995 for the evaluation of medicinal products.
Suspected ADRs are submitted by the health care professionals and patients to EudraVigilance or MAHs. The Pharmacovigilance Risk Assessment Committee evaluates the total risk perspectives and supervision of drugs. The above committee incorporates the findings, assessment, reduction, and announcement concerning the hazards of adverse reactions while keeping the remedial outcome of the drug in consideration.
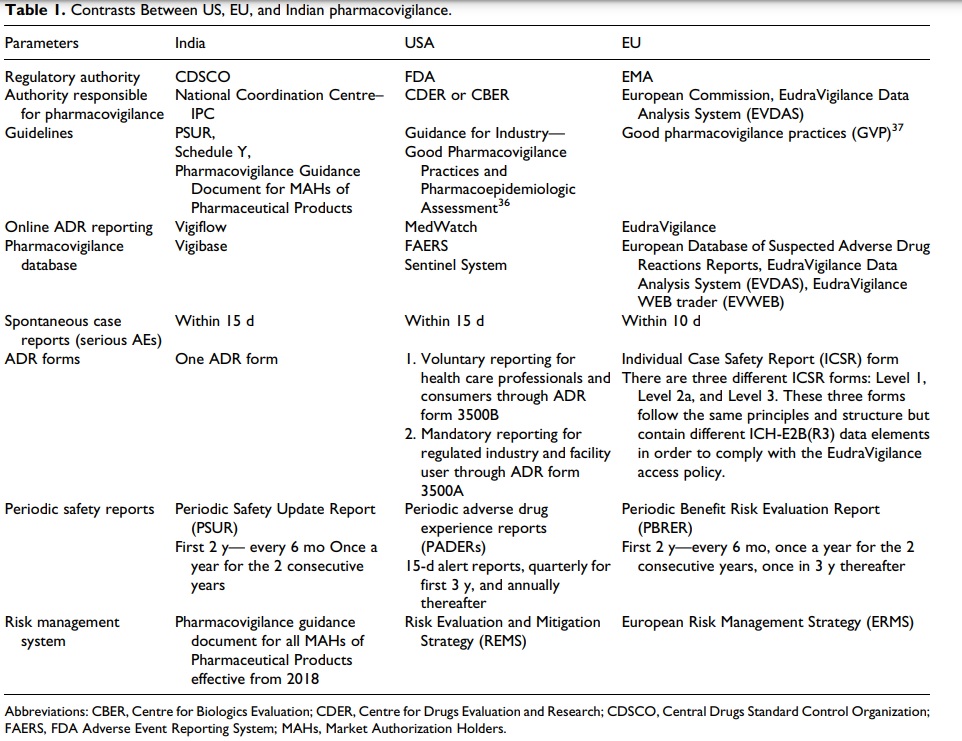
Contrasts Between US, EU, and Indian PV
PV is still in a nascent stage in India compared with the PV system in both EU and USA. Both the countries require obligatory reporting of all serious adverse events, whereas in India there were no particular guidelines for reporting of ADR other than Schedule Y until PvPI came into place.
Recently in 2018, it became obligatory for the MAHs to report ADRs under PvPI. MedWatch and EudraVigilance are the web-based ADR reporting systems of USA and EU, respectively, while India
uses the WHO-based Vigiflow system. The ADR forms are not classified into two different types as in the US and EU systems.