Parenterals product requiring sterile packaging: Production facilities and MCQs for GPAT, NIPER, Pharmacist and Drug Inspector exam
The production facility and its associated equipment must be designed, constructed, and operated properly for the manufacture of a sterile product to be achieved at the quality level required for safety and effectiveness. The processes used must meet cGMP standards.
Functional areas: To achieve the goal of a manufactured sterile product of exceptionally high quality, many functional production areas are involved: warehousing or procurement; compounding (formulation); materials (containers, closures, equipment) preparation; filtration and sterile receiving; aseptic filling; stoppering; lyophilization (if warranted); and packaging, labeling, and quarantine. The extra requirements for the aseptic area are designed to provide an environment where a sterile fluid may be exposed to the environment for a brief period during subdivision from a bulk container to individual-dose containers, without becoming contaminated. Contaminants, such as dust, lint, and other particles and micro-organisms, are found floating in the air, lying on counters and other surfaces, attached to clothing and body surfaces of personnel, concentrated in the exhaled breath of personnel, and deposited on the floor. The design and control of an aseptic area is directed toward reducing the presence of these contaminants, so they are no longer a hazard to aseptic filling.
Flow Plan: In general, the components for a parenteral product flow from the warehouse, after release, to either the compounding area, as for ingredients of the formula, or the materials support area, as for containers and equipment. After proper processing in these areas, the components flow into the security of the aseptic area for filling of the product in appropriate containers. From there the product passes into the quarantine and packaging area, where it is held until all necessary tests have been performed. If the product is to be sterilized in its final container, its passage is interrupted after leaving the aseptic area for subjection to the sterilization process. After the results from all tests are known, the batch records have been reviewed, and the product has been found to comply with its release specifications, it passes to the finishing area for final release for shipment.
Clean room classified areas: Due to the extremely high standards of cleanliness and purity that must be met by parenteral products, it has become standard practice to prescribe specifications for the environments (clean rooms) in which these products are manufactured.
Table 1 – Clean room classifications
| European grade | United States classification | International Society of Pharm. Eng. description | Max. no. of particles (per m3>/=0.5 μm) | Max. no. of particles (per m 3 >/= 5 μm) |
| A | 100 | Critical | 3,500 | 0 |
| B | 100 | Clean | 3,500 | 0 |
| C | 10,000 | Controlled | 350,000 | 2,000 |
| D | 100,000 | Pharmaceutical | 3,500,000 | 20,000 |
The classifications used in pharmaceutical practice normally range from Class 100,000 (Grade D) for materials support areas to Class 100 (Grade A) for aseptic areas. To achieve Class 100 conditions, HEPA filters are required for the incoming air, with the effluent air sweeping the downstream environment at a uniform velocity, 100 ft/min ± 20%, along parallel lines (laminar air flow). HEPA filters are defined as 99.99% or more efficient in removing, from the air, 0.3 μm particles generated by vaporization of the hydrocarbon Emory 3004.
European standards differ from US standards, as European standards:
- use Grades A, B, C, and D classifications, rather than Class X (100, 1,000, etc);
- use particle and microbial limits per cubic meter, rather than per cubic foot;
- require particle measurements at 5 microns in addition to 0.5 microns in Grade A and B areas; and
- differentiate area cleanliness dynamically and ‘at rest.’
Air Cleaning—Since air is one of the greatest potential sources of contaminants in clean rooms, special attention must be given to air drawn into clean rooms by the heating, ventilating, and air conditioning (HVAC) systems. For personnel comfort, air conditioning and humidity control should be incorporated into the system. The clean, aseptic air is introduced into the Class 100 area and maintained under positive pressure, which prevents outside air from rushing into the aseptic area through cracks, temporarily open doors, or other openings.
Laminar-Flow Enclosures—The required environmental control of aseptic areas has been made possible by the use of laminar airflow, originating through a HEPA filter, occupying one entire side of the confined space. Therefore, it bathes the total space with very clean air, sweeping away contaminants. The orientation for the direction of airflow can be horizontal or vertical and may involve a limited area, such as a workbench, or an entire room.
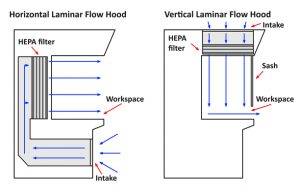
Figure 1 – Horizontal and vertical laminar air flow
Materials Support Area—This area is constructed to withstand moisture, steam, and detergents and is, usually, a Class 100,000 clean room. The ceiling, walls, and floor should be constructed of impervious materials, so moisture runs off and is not held. One of the finishes with a vinyl or epoxy sealing coat provides a continuous surface free from all holes or crevices. All such surfaces can be washed at regular intervals to keep them thoroughly clean. These areas should be exhausted adequately, so the heat and humidity are removed for the comfort of personnel. Precautions must be taken to prevent the accumulation of dirt and the growth of micro-organisms due to the high humidity and heat. In this area, preparation for the filling operation, such as cleaning and assembling equipment, is undertaken. Adequate sink and counter space must be provided. This area must be cleanable, and the microbial load must be monitored and controlled. Precautions must also be taken to prevent deposition of particles or other contaminants on clean containers and equipment, until they have been properly boxed or wrapped preparatory to sterilization and depyrogenation.
Compounding Area—The formula is compounded in this area. Although it is not essential that this area be aseptic, control of micro-organisms and particulates should be more stringent than in the materials support area. For example, means may need provided to control dust generated from weighing and compounding operations. Cabinets and counters should, preferably, be constructed of stainless steel. They should fit snugly to walls and other furniture, so there are no catch areas dirt can accumulate. The ceiling, walls, and floor should be similar to those for the materials support area.
Aseptic Area—The aseptic area requires construction features designed for maximum microbial and particulate control. The ceiling, walls, and floor must be sealed, so they may be washed and sanitized with a disinfectant, as needed. All counters should be constructed of stainless steel and hung from the wall, so there are no legs to accumulate dirt, where they rest on the floor. All light fixtures, utility service lines, and ventilation fixtures should be recessed in the walls or ceiling to eliminate ledges, joints, and other locations for the accumulation of dust and dirt. As much as possible, tanks containing the compounded product should remain outside the aseptic filling area, with the product fed into the area through hose lines. Proper sanitization is required, if the tanks must be moved in. Large mechanical equipment located in the aseptic area should be housed as completely as possible within a stainless steel cabinet, to seal the operating parts and their dirt-producing tendencies from the aseptic environment. Further, all such equipment parts should be located below the filling line. Mechanical parts that will contact the parenteral product should be demountable, so they can be cleaned and sterilized. Personnel entering the aseptic area should enter only through an airlock. They should be attired in sterile coveralls with sterile hats, masks, goggles, foot covers, and double gloves. Movement within the room should be minimal, and in-and-out movement rigidly restricted during a filling procedure. The requirements for room preparation and the personnel may be relaxed, if the product is to be sterilized terminally in a sealed container. Some are convinced, however, it is better to have one standard procedure meeting the most rigid requirements.
Isolation (Barrier) Technology—Isolator (or barrier) technology has long been used in the pharmaceutical industry and ranges from simple screens to restricted access barriers (RABS) to full isolation systems, all designed to isolate aseptic operations from personnel and the surrounding environment. Sterility tests are now almost exclusively conducted within isolators. Isolators are enclosed, usually positively pressurized units with high efficiency particulate air (HEPA) filters, supplying ISO 5 airflow in a unidirectional manner to the interior. Air recirculates by returning it to the air handlers through sealed ductwork. Cleaning can be manual or automated (clean-inplace). Access to an isolator is through glove ports and sterile transfer systems. Isolators can be located in an ISO 8 or better environment. The operations are performed within windowed, sealed walls, with operators working through glove ports. The sealed enclosures are presterilized, usually with peracetic acid, hydrogen peroxide vapor, or steam. Sterile supplies are introduced from sterilizable, movable modules, through uniquely engineered transfer ports or directly from attached sterilizers, including autoclaves and hot-air sterilizing tunnels. Several factors have contributed to the increased importance and utilization of barrier isolator technology:
- The high level of concern from manufacturers and regulatory agencies over the level of sterility assurance in aseptic processing.
- Continued, relatively high level of product recalls, due to concerns—proven or suspected—over contamination potential.
- The surge of potential heat-labile products from biotechnology and the inability to terminally sterilize these molecules. There are needs to control the environment not only from contamination, but also with respect to stability considerations—temperature, humidity, and, if necessary, anaerobics.
- Many new drug compounds are cytotoxic or otherwise highly potent, where safety considerations demand separation of these drugs from human operators.
- Because so many biopharmaceutical drugs are so expensive, there is a trend toward smaller batch production. Smaller batch production makes construction of large manufacturing facilities unnecessary, yet there is still the need to manufacture in Class 100/Grade A/ISO 5 clean rooms. Isolators are ideal for smaller facilities, plus are much more economical from the standpoint of capital, labor and maintenance, and operator (e.g., number of employees, gowning) costs.
Maintenance of clean rooms:
- Assuming the design of the facilities is cleanable and sanitizable, a carefully planned cleaning schedule should be developed, ranging from daily to monthly, depending on the location and its relation to the most critical Class 100 areas. Tools used should be non-linting, designed for clean room use, held captive to the area, and, preferably, sterilizable.
- Liquid disinfectants (sanitizing agents) should be selected carefully, due to data showing their reliable activity against inherent environmental micro-organisms. They should be recognized as supplements to good housekeeping, never as substitutes. They should be rotated with sufficient frequency to avoid the development of resistant strains of micro-organisms.
- It should be noted that ultraviolet (UV) light rays of 237.5 nm wavelength, as radiated by germicidal lamps, are an effective surface disinfectant. It is stated that an irradiation intensity of 20 μw/cm2 is required for effective antibacterial activity.
Personnel:
- Personnel selected to work on the preparation of a parenteral product must be neat, orderly, and reliable.
- They should be in good health and free from dermatological conditions that might increase the microbial load. If personnel show symptoms of a head cold, allergies, or similar illness, they should not be permitted in the aseptic area, until recovery is complete.
- Aseptic-area operators should be given thorough, formal training in the principles of aseptic processing and the techniques to be employed.
- The uniform worn is designed to confine the contaminants discharged from the body of the operator, thereby preventing their entry into the production environment. Aseptic area, uniforms should be sterile. Fresh, sterile uniforms should be used after every break period or whenever the individual returns to the aseptic area.
- Uniforms, usually, consist of coveralls for both men and women, hoods to cover the hair completely, face masks, and Dacron or plastic boots. Sterile rubber or latexfree gloves are also required for aseptic operations, preceded by thorough scrubbing of the hands with a disinfectant soap. Two pairs of gloves are put on, one pair at the beginning of the gowning procedure, the other pair after all other apparel has been donned. In addition, goggles are required to complete the coverage of all skin areas.
Environmental control evaluation: Manufacturers of sterile products use extensive means to control the environment, so these critical injectable products can be prepared free from contamination. Nevertheless, tests should be performed to determine the level of control actually achieved. Normally, the tests consist of counting viable and non-viable particles suspended in the air or settled on surfaces in the workspace. The tests used measure either the particles in a volume of sampled air or the particles settling or present on surfaces. To measure the total particle content in an air sample, electronic particle counters are available, operating on the principle of the measurement of light scattered from particles as they pass through the cell of the optical system. Locations for sampling should be planned to reveal potential contamination levels that may be critical in the control of the environment. The sample should be large enough to obtain a meaningful particle count.
The slit-to-agar (STA) sampler draws, by vacuum, a measured volume of air through an engineered slit, causing the air to impact the surface of a slowly rotating nutrient agar plate. Micro-organisms adhere to the surface of the agar and grow into visible colonies counted as CFUs, since it is not known whether the colonies arise from a single micro-organism or a cluster.
A widely used method for microbiological sampling consists of the exposure of nutrient agar culture plates to the settling of micro-organisms from the air. This method is very simple and inexpensive to perform, but will detect only those organisms that have settled on the plate; therefore, it does not measure the number of micro-organisms in a measured volume of air (a non-quantitative test).
Media fill (Process simulation testing): FDA inspections have increasingly focused on media fill studies that truly simulate the production process. The media fill or process simulation test involves preparation and sterilization (often by filtration) of sterile trypticase soy broth and filling sterile containers with this broth, under conditions simulating, as closely as possible, those characteristics of a filling process for a product. The key is designing these studies to simulate all factors that occur during the normal production of a lot. Media fills are conducted, when a new filling line or new product container is introduced. All personnel involved in the aseptic filling of a product (i.e., operators, maintenance personnel, microbiology support personnel) must participate in at least one media fill run per year. The culture media used for each media fill exercise must be tested to ensure it will support the growth of micro-organisms, if they are present.
Table 2 – Factors to consider in the design of media fill studies
| · Duration of longest run |
| · Worst – case environmental conditions |
| · Number and type of interventions, stoppages, adjustments, transfers |
| · Aseptic assembly of equipment |
| · Number and activities of personnel |
| · Number of aseptic conditions |
| · Shift breaks, changes, multiple gownings |
| · Number/type of aseptic equipment disconnections and connections |
| · Aseptic samples |
| · Line speed/configuration |
| · Manual weight checks |
| · Operator fatigue |
| · Container/closure types run on the line |
| · Temperature/relative humidity extremes |
| · Conditions permitted before line clearance |
| · Container/closure surfaces which contact formulation during aseptic process |
Other requirements of a valid media fill experiment include:
- Must have the appropriate criteria for batch yield and accountability, just like a product batch.
- Must identify any contaminant to the species level and perform complete investigations of failed media fills.
- FDA advocates videotape media fills to identify personnel practices that could negatively impact the aseptic process.
- Media fill duration, according to FDA, EU, ISO, CEN (European Committee of Standardization), and PIC, must be sufficiently long to include all required manipulations and cover the same length of time normally consumed by the commercial process. Most media fills are a minimum of 3 hours; some may be as long as 24 hours.
Multiple choice questions:
1.The production facility and its associated equipment must be designed, constructed, and operated properly for the manufacture of a sterile product to be achieved at the quality level required for safety and effectiveness. The processes used must meet ____ standards.
a)GLP
b)GMP
c)cGMP
d)All of these
2.To achieve the goal of a manufactured sterile product of exceptionally high quality, many functional production areas are involved like
a)warehousing
b)compounding
c)filtration
d)all of these
3.The design and control of an aseptic area is directed toward reducing the presence of these contaminants, so they are no longer a hazard to aseptic filling.
a)true
b)false
4.Clean room classifications European grade is/are
a)A
b)B
c)C
d)All of these
5.European grade A area is
a)class 100
b)class 1000
c)class 10000
d)class 100000
6.Class 100 area is
a)Critical area
b)Controlled area
c)Pharmaceutical area
d)All of these
7.To achieve Class 100 conditions, _____ are required
a)Membrane filters
b)HEPA filters
c)Seitz filters
d)Proper ventilation
8.HEPA filters are defined as _____% or more efficient in removing, from the air, 0.3 μm particles generated by vaporization of the hydrocarbon Emory 3004.
a)10
b)50
c)70
d)99.99
9.European standards differ from US standards, as European standards
a)use Grades A, B, C, and D classifications, rather than Class X (100, 1,000, etc)
b)use particle and microbial limits per cubic meter, rather than per cubic foot
c)require particle measurements at 5 microns in addition to 0.5 microns in Grade A and B areas
d)all of these
10.HVAC stands for
a)heating, ventilating, and air conditioner
b)heating, ventilating, and air conditioning
c)heat, ventilating, and air conditioning
d)heating, ventilator, and air conditioning
11.Materials Support Area is constructed to withstand
a)moisture
b)steam
c)detergents
d)all of these
12.It should be noted that ultraviolet (UV) light rays of ____ nm wavelength, as radiated by germicidal lamps, are an effective surface disinfectant.
a)237.5
b)230.5
c)235
d)240
13.It is stated that an irradiation intensity of 20 μw/cm2 is required for effective antibacterial activity.
a)true
b)false
14.Media fills are conducted, when
a)a new filling line is introduced
b)a new product container is introduced
c)an old product is reintroduced
d)a and b
15.Factors to consider in the design of media fill studies is/are
a)Duration of longest run
b)Aseptic assembly of equipment
c)Number of aseptic conditions
d)All of these
Solutions:
- c)cGMP
- d)all of these
- a)true
- d)All of these
- a)class 100
- a)Critical area
- b)HEPA filters
- d)99.99
- d)all of these
- b)heating, ventilating, and air conditioning
- d)all of these
- a)237.5
- a)true
- d)a and b
- d)All of these
References:
- Remington Essential of Pharmaceutics, 1st edition 2013, page no. 510-517.
List of Successful GPATINDIAN CANDIDATES
Participate in Online FREE GPAT TEST: CLICK HERE
Participate in Online FREE Pharmacist TEST: CLICK HERE
Participate in Online FREE Drug Inspector TEST: CLICK HERE
Participate in CSIR NET JRF Mock Test